Our client sought to conduct a covalent fragment screen to discover selective covalent inhibitors targeting a Mg-dependent metalloenzyme. During initial assay optimisation, various compound classes known to cause interference in certain assays appeared to inhibit function of the target. Following the primary screen, several hits failed to reconfirm after resynthesis, despite initially appearing to act as inhibitors.
Careful assay development and optimisation protected our client from pursuing misleading false positives, preventing months of unnecessary downstream effort, wasted resources and avoidable costs. As a result of the co-location of BioAscent’s integrated teams, compound resynthesis, assay troubleshooting, and hypothesis testing were significantly accelerated compared with outsourcing chemistry and biosciences to separate CROs.
Covalent fragment screening is becoming an increasingly popular hit-finding technique in drug discovery, offering a promising way to target challenging proteins with high specificity. It is particularly valuable for proteins considered “undruggable” with traditional reversible small molecules.
Irreversible covalent enzyme inhibitors act through a two-step mechanism (Figure 1)1:
Reversible non-covalent recognition:
The inhibitor binds to the target protein through typical molecular interactions governed by kon and koff (for example, hydrogen bonds or hydrophobic contacts). This ensures that the reactive “warhead” is positioned near a specific nucleophilic amino acid residue. This step is essential for selectivity and minimising off target effects.
Irreversible covalent bond formation:
The electrophilic warhead forms a covalent bond with the target residue and, in the case of irreversible covalent inhibitors, permanently modifies the protein until it is degraded. This process is described by the first-order rate constant, kinact.
This covalent bond formation step results in sustained inhibition even after the free drug has cleared from circulation, meaning covalent inhibitors can achieve high potency and reduced dosing frequency.
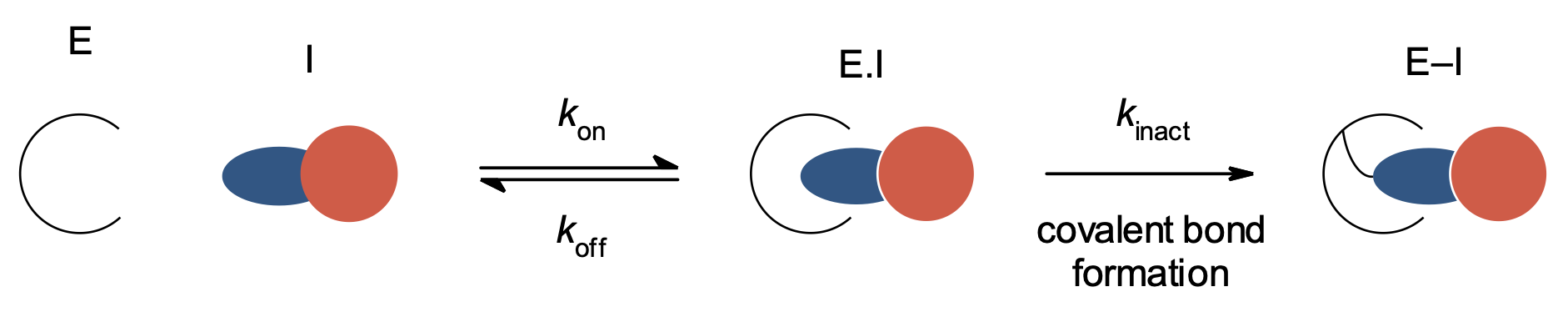 Figure 1. Mechanism of irreversible enzyme inhibitors. E = enzyme, I = inhibitor; inhibitor warhead is shown in blue.
Figure 1. Mechanism of irreversible enzyme inhibitors. E = enzyme, I = inhibitor; inhibitor warhead is shown in blue.
The client, a venture-backed biotech developing first-in-class therapeutics, sought to run a covalent fragment screen to identify selective covalent inhibitors for a Mg-dependent metalloenzyme that could provide high quality starting points for medicinal chemistry.
Covalent fragment screening demands robust assay development and a carefully considered cascade of assays to ensure the accurate detection and validation of covalent binders. Non-specific interactions can be prevalent, therefore in-depth data analysis is crucial to separate true hits from false positives.
Assay development
BioAscent scientists developed a robust colorimetric assay using BIOMOL® Green for phosphate quantification in a microplate format to measure the activity of the Mg-dependent bi-substrate metalloenzyme target (Figure 2).
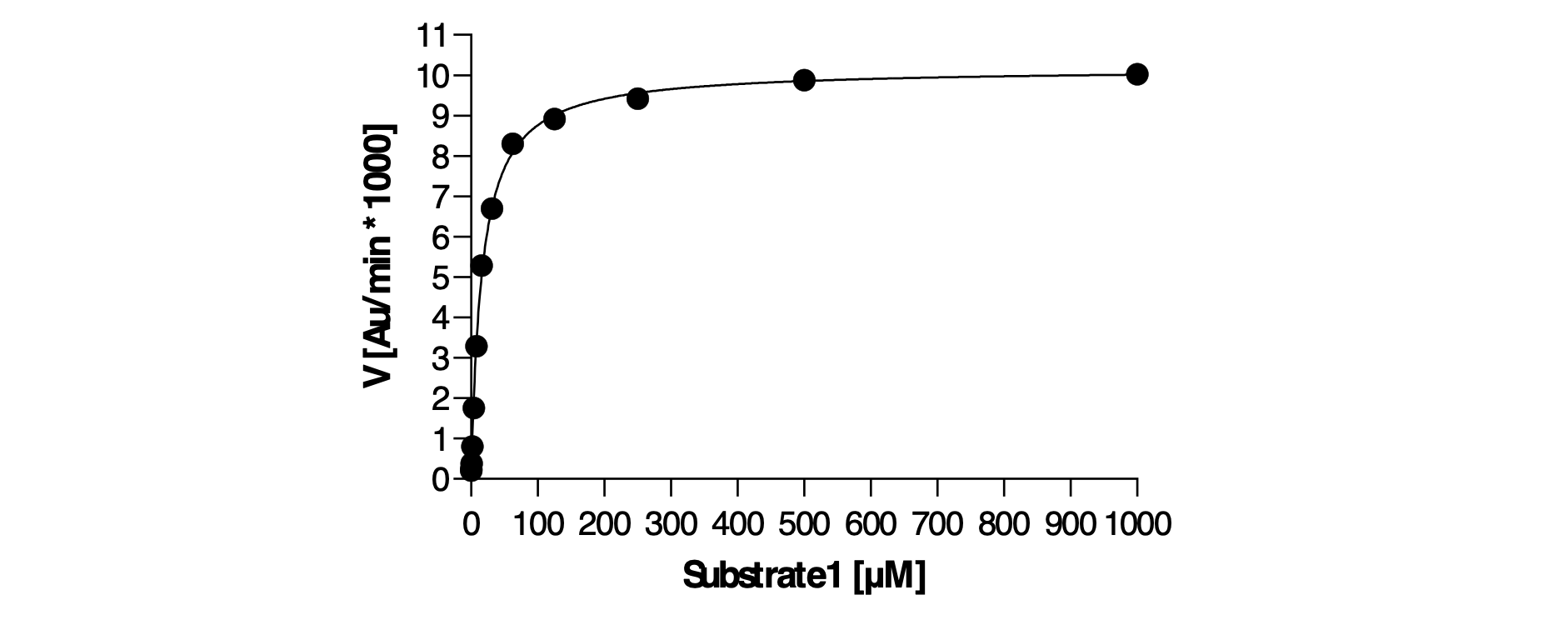 Figure 2. Assay optimisation included setting a low nM enzyme and steady-state substrate concentrations and reaction time, ensuring a robust and sensitive assay.
Figure 2. Assay optimisation included setting a low nM enzyme and steady-state substrate concentrations and reaction time, ensuring a robust and sensitive assay.
The assay buffer was then optimised to maintain enzyme stability and activity while minimising interference. BioAscent’s proprietary Pan-Assay Interference (PAINS) library was tested under initial buffer conditions, showing that the assay was susceptible to inhibition by various known interference compounds and common contaminants of synthesis, particularly divalent cations. Optimisation of the buffer reduced interference, though some inhibition by divalent cations remained (Figure 3).
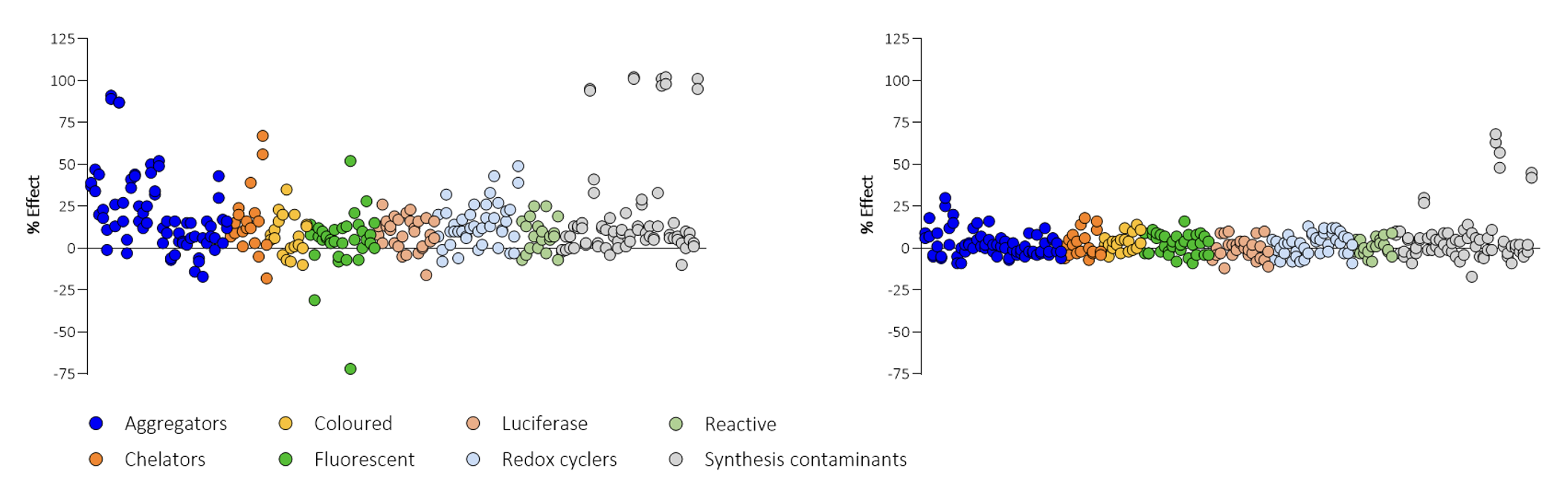 Figure 3. Initially, the assay was inhibited by various compounds, including aggregators, chelators, redox cycling compounds and divalent cations (left). The buffer was modified by optimising the composition and concentration of detergent, reducing agent, carrier proteins and the introduction of mild chelating agents, which dramatically reduced assay interference (right), although some inhibition by divalent cations remained.
Figure 3. Initially, the assay was inhibited by various compounds, including aggregators, chelators, redox cycling compounds and divalent cations (left). The buffer was modified by optimising the composition and concentration of detergent, reducing agent, carrier proteins and the introduction of mild chelating agents, which dramatically reduced assay interference (right), although some inhibition by divalent cations remained.
Compound screening
A covalent library of 1600 compounds was sourced from a commercial third party and screened in the primary assay at 100 µM. Enzyme activity was assessed at steady-state and from 77 hits identified in the initial single-point screen, 38 showed time-dependent inhibition (Figure 4).
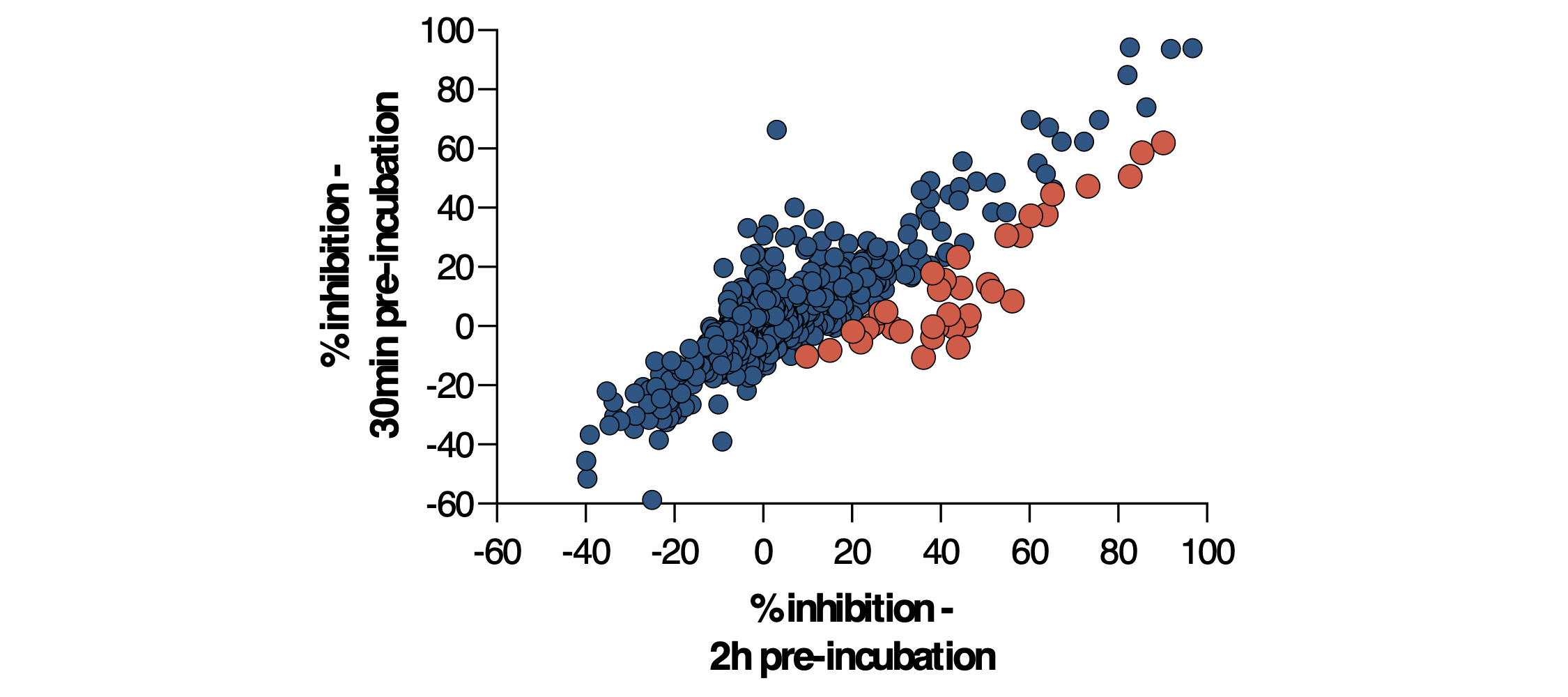 Figure 4. 77 compounds were flagged as primary hits from the 30-minute incubation screen. Of these, 38 appeared to show time-dependent inhibition (shown in orange).
Figure 4. 77 compounds were flagged as primary hits from the 30-minute incubation screen. Of these, 38 appeared to show time-dependent inhibition (shown in orange).
Promising hits were re-synthesised by the medicinal chemistry team at BioAscent. While some compounds confirmed the activity, many were found to be inactive (Figure 5A), prompting a deeper investigation. Since divalent cations were known to impact assay performance, the assay buffer was reoptimised to include a higher concentration of chelator while retaining enzyme activity. Several screening samples lost activity in the presence of 50 mM EGTA (Figure 5B), indicating that their initial activity arose from heavy metals in the sample and not from meaningful target engagement. After identifying this non-specific inhibition caused by contaminants, 113 compounds of interest were re-screened in a new buffer, with 15 hits retaining clear, time-dependent inhibition.
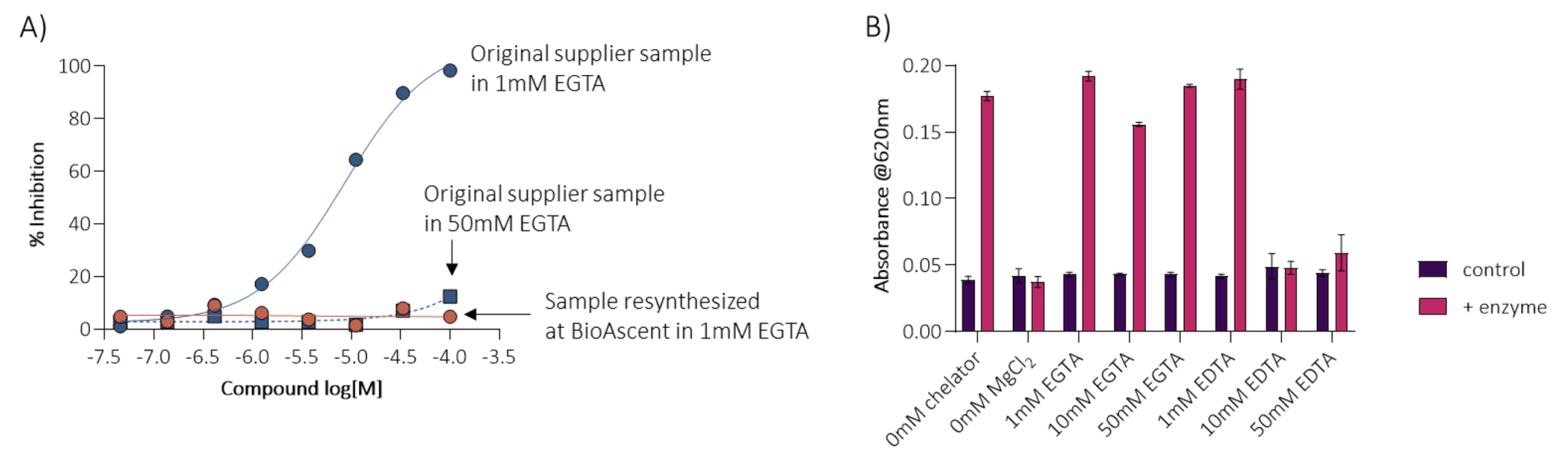 Figure 5A. Hit re-synthesised by the BioAscent chemistry team was inactive when compared to the original sample; Figure 5B. The buffer was reoptimised to include a higher concentration of chelator, while retaining enzyme activity.
Figure 5A. Hit re-synthesised by the BioAscent chemistry team was inactive when compared to the original sample; Figure 5B. The buffer was reoptimised to include a higher concentration of chelator, while retaining enzyme activity.
Compound characterisation
Confirmed inhibitors from the primary screen were further profiled using the BIOMOL® Green assay to elucidate their mechanism of action and guide a medicinal chemistry programme focused on understanding structure-activity relationships (SAR).
Compounds were first tested in an IC50 shift assay (Figure 6). A significant IC50 shift indicated time-dependent covalent binding and enabled calculation of kinact/KI, a key metric for assessing covalent inhibition efficiency.2,3
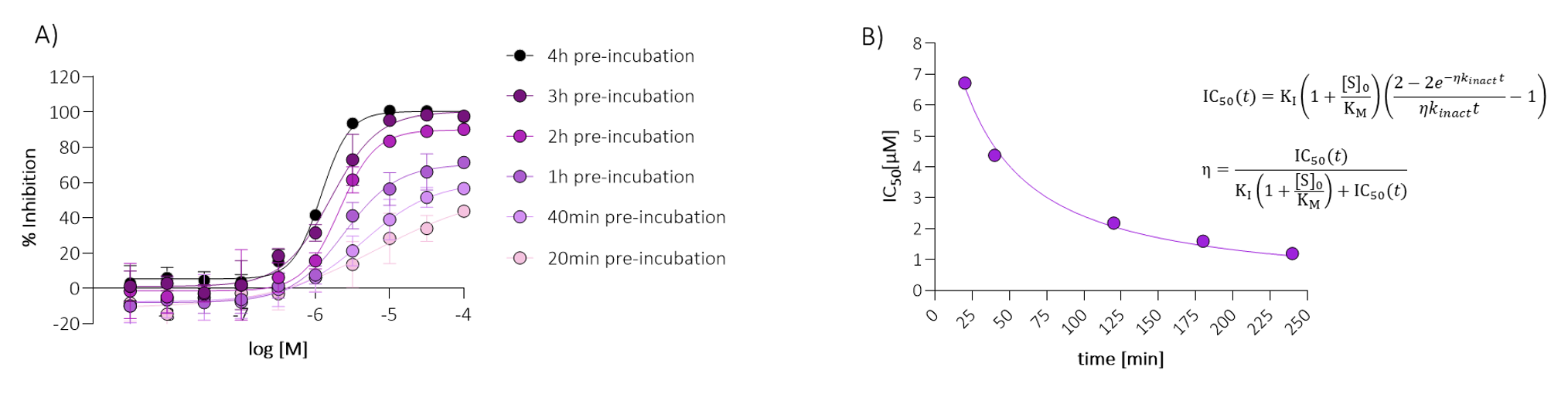 Figure 6. The IC50 shift assay.
Figure 6. The IC50 shift assay.
Compounds were then profiled in a jump dilution assay, where persistent inhibition after dilution indicated irreversible binding with the target (Figure 7). Subsequent mode of action studies provided further insight into the mode of inhibition for each confirmed hit, providing vital information for SAR (Figure 8).
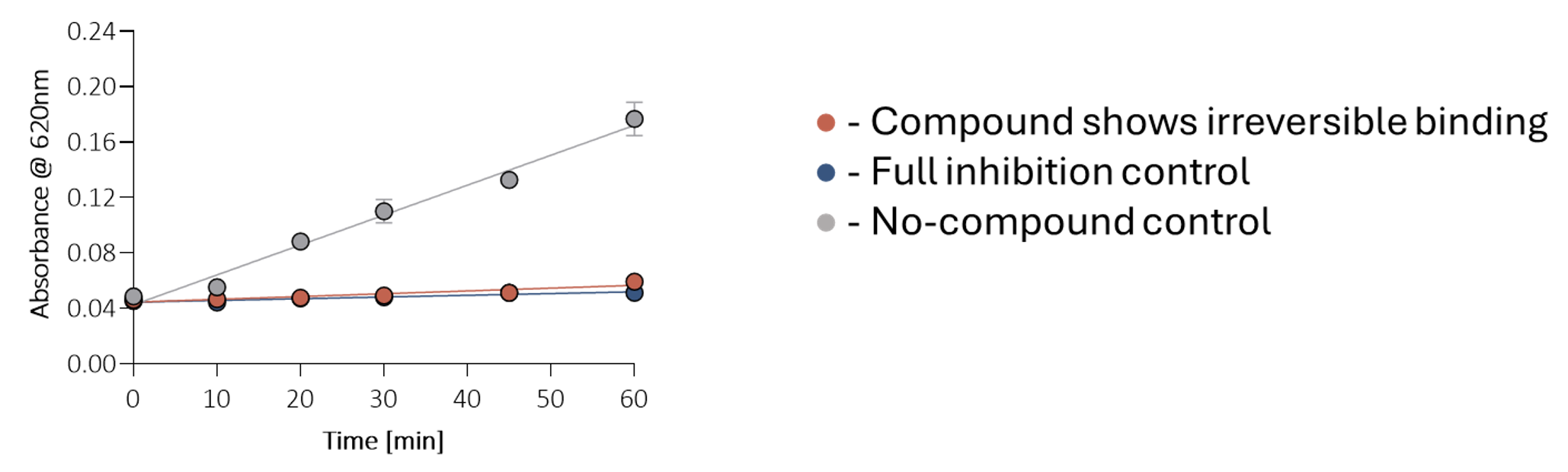 Figure 7. The jump dilution assay. The inhibitor is pre-incubated at 10xIC50 concentration with 100xFAC enzyme concentration, then diluted 1:100 into the active assay, and enzyme activity is monitored over time. Persistent inhibition after dilution indicates irreversible binding. Fully inhibited and no-compound controls were included for comparison in each experiment.
Figure 7. The jump dilution assay. The inhibitor is pre-incubated at 10xIC50 concentration with 100xFAC enzyme concentration, then diluted 1:100 into the active assay, and enzyme activity is monitored over time. Persistent inhibition after dilution indicates irreversible binding. Fully inhibited and no-compound controls were included for comparison in each experiment.
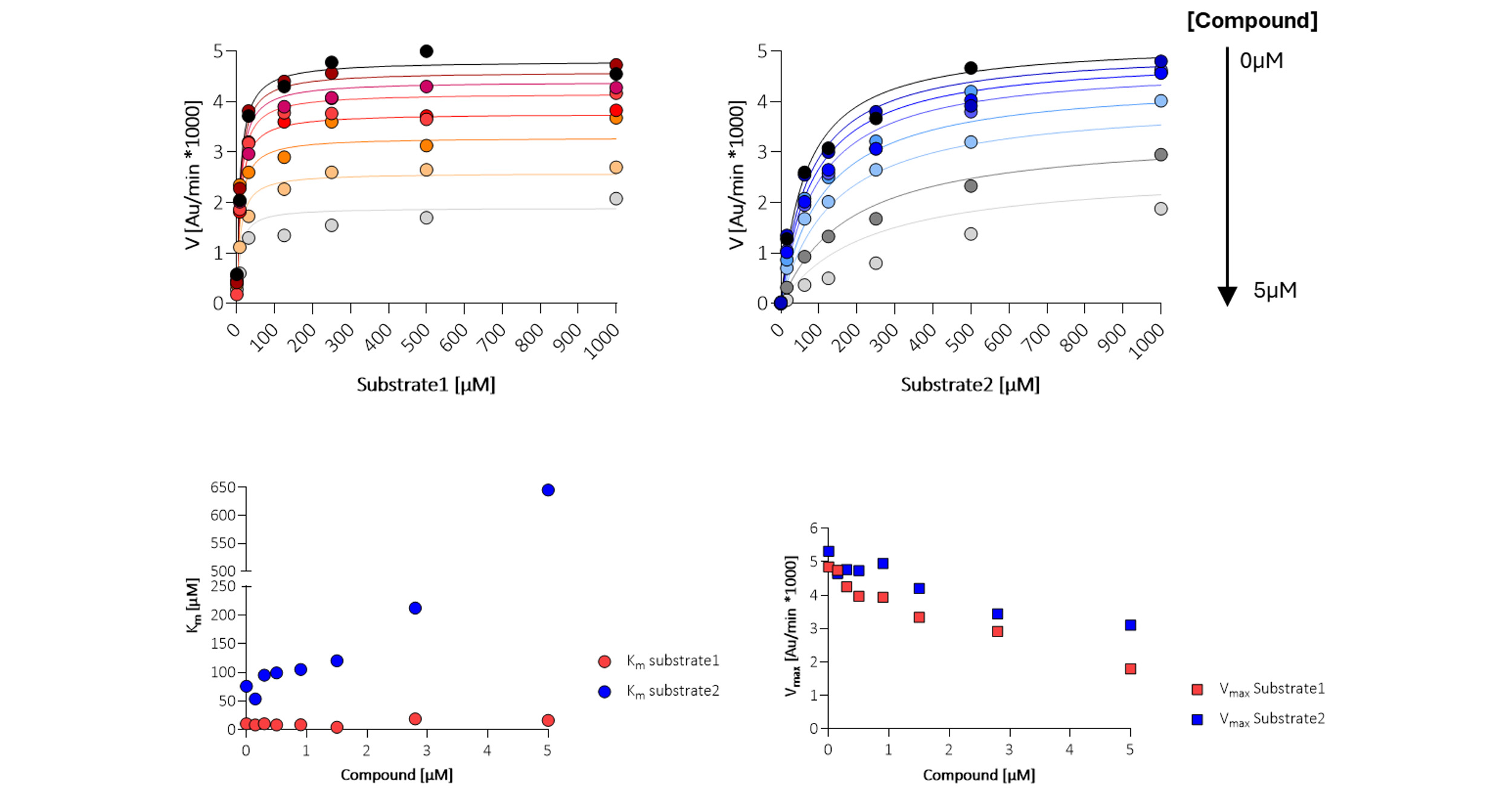 Figure 8. Mode of action studies. Each confirmed inhibitor was analysed for effect on KM (Michaelis constant) and Vmax (maximum velocity) to determine the mode of inhibition, assuming a constant a. The tested compound shows competitive inhibition (blue) for substrate 2 and non-competitive inhibition (red) for substrate 1.
Figure 8. Mode of action studies. Each confirmed inhibitor was analysed for effect on KM (Michaelis constant) and Vmax (maximum velocity) to determine the mode of inhibition, assuming a constant a. The tested compound shows competitive inhibition (blue) for substrate 2 and non-competitive inhibition (red) for substrate 1.
Buffer optimisation during initial assay development was essential to minimise the target’s sensitivity to interference compounds. Despite this optimisation, some hits could not be re-confirmed after resynthesis, even though they initially appeared to be active. Further analysis revealed that high concentrations of chelators were necessary to restore enzyme activity, indicating that significant metal contamination was present in some library samples.
This finding was critical as without it, several false positives may have progressed into downstream chemistry despite having no genuine inhibitory activity. Eliminating these misleading results saved the client months of unnecessary effort.
The co‑located integrated chemistry and biology teams at BioAscent enabled rapid compound resynthesis, immediate assay re‑evaluation, and fast, cohesive problem‑solving; an approach that significantly shortened the cycle of analysis and troubleshooting compared with outsourcing chemistry activities to a separate CRO.
Extensive biochemical characterisation of genuine hit compounds by our team provided the confidence required for the client to advance into medicinal chemistry with the well-validated hit series identified from the screen. The work ultimately identified selective covalent inhibitors that served as chemically tractable starting points, positioning the client for rapid progression into hit-to-lead development.
Learn more about BioAscent’s integrated drug discovery expertise here.
The work in this case study was performed by BioAscent Senior Scientists Dominika Kowalczyk and Michael Mathieson.
Dominika is a bioscientist with extensive experience working across assay development, structural biology and project leadership. Michael is an experienced medicinal chemist specialising in organic synthesis and small molecule drug design.

Heppner, D. et al., J. Med. Chem. 2024, 67, 14693. https://doi.org/10.1021/acs.jmedchem.4c01721
Mons, E. et al., Current Protocols, 2022, 2, e419. https://doi.org/10.1002/cpz1.419
Huisinga, W. et al., SLAS Discovery, 2009, 14(8), 913. https://doi.org/10.1177/1087057109336751




